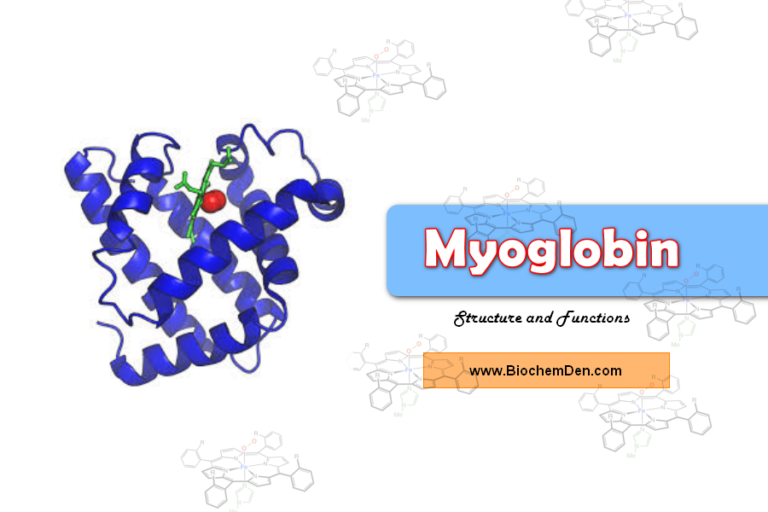
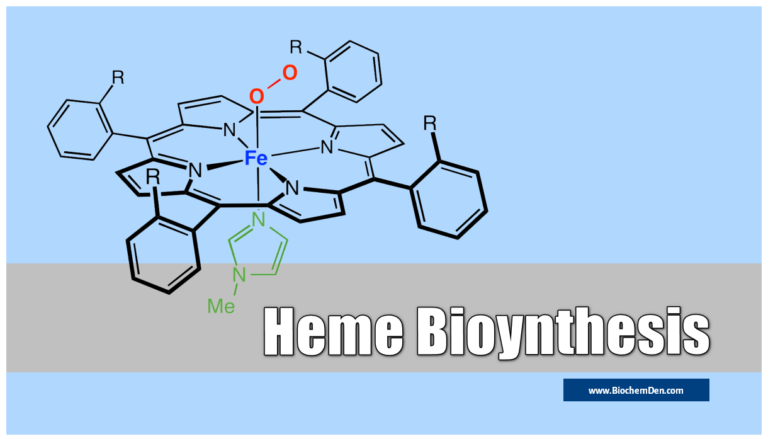
Hemoglobin is the main part of red blood cells. These cells help move oxygen around the body and control how much oxygen gets to different tissues. Hemoglobin cannot be produced in the body alone and is produced and stored when the amount of hemoglobin in the body is maintained at a steady level.
The average person has a hemoglobin count of 14.8 grams per 100 ml of blood. Let us check the details of what is the structure and function of hemoglobin. Let us describe hemoglobin structure and functions in this article.
What is Hemoglobin?
Hemoglobin is a globular heme protein in vertebrate red blood cells and in the plasma of many invertebrates that carries oxygen and carbon dioxide. The heme group binds oxygen and carbon dioxide and imparts a red color to the blood; it is also spelled hemoglobin.

- Why Proteins are Very Important? How to Explain?
- Peptide bonds: Backbone of the Proteins
- What is Amino acid and its Structural Chemistry?
Historical Aspects of Hemoglobin Discovery
- In 1665, Marcello Malpighi described the RBCs.
- Felix Hope Seyler, in 1862, isolated pure hemoglobin.
- In 1904, Christian Bohr discovered that hemoglobin is the transporter of oxygen.
- In 1912, Kutster established the structure of hemoglobin.
- Hans Fischer synthesized heme in the laboratory in 1920 (Nobel Prize, 1930).
- In 1945, Linus Pauling (Nobel Prize, 1954) described abnormal hemoglobins.
- Max Perutz (Nobel Prize, 1962) studied the 3D structure of hemoglobin.
Structure of Hemoglobin
Let me explain the structure of hemoglobin. Here are the details of the hemoglobin structure and functions.
- Heme is an iron porphyrin compound. Porphyrin is a tetrapyrrole structure.
- Ferrous iron occupies the center of the porphyrin ring and establishes linkages with all four nitrogens of all the pyrrole rings.
- It is also linked to the nitrogen of the imidazole ring of histidine present in the globin part.
- The globin part comprises four polypeptide chains, two identical polypeptide chain in hemoglobin. They are α-chains, and two identical β-chains in normal adult hemoglobin.
- Each chain contains a “heme” in the so-called ‘heme pocket.’ So one Hb molecule possesses four heme units.
- The Hb molecule contains hydrophobic amino acids and hydrophilic ones on the surface.
- Heme pockets of α-subunits are just the adequate size to allow entry of an O2 molecule. Entry f O2 into heme pockets of β-subunits is blocked by a valine residue.

- The composition of hemoglobin is having the following,
- Hb-A1 (two α-chains and β-chains)
- HbF (two α-chains and ¥-chains)
- Hb-A2 (two α-chains and delta-chains)
- Embryonic Hb (two α-chains and €-chains)
- Hb-A3 (altered from Hb-A found in old red cells)
- HbA1C (glycosylated Hb, present in a concentration of 3–5% of total Hb). In diabetes mellitus, it is increased to 6 to 15%.
What is the hemoglobin formula structure?
- The formula for hemoglobin can be written as C₈₈H₁₄₈O₂₂N₈Fe.
- It represents the chemical composition of hemoglobin, composed of four protein subunits, each containing a heme group with an iron atom in the center.
- The iron atom is responsible for binding to oxygen molecules and allowing oxygen transport in the bloodstream.
Globin Proteins
- A red-colored conjugated protein (made up of heme and Globin) is present inside the RBC.
- Normal Hb% in an adult male is 14 to 16 gm.
- Approximately 6.25 gm of Hb is synthesized and destroyed every day.
- Heme structure does not vary from species to species.
- The essential protein globin varies in amino acid composition and sequence in different species.
- Globin is rich in Histidine and lysine.
- it gives the basic structure of haemoglobin.
Functions of haemoglobin
The functions of hemoglobin biomolecule is given below
- Hb binds O2, transports O2, and delivers the same to tissues.
- Hb binds CO2, a waste product of metabolism.
- 2-3 BPG, produced in RBC by Rapport-Leubering shunt, stabilizes Hb confirmation at the quaternary level and enhances dissociation of O2 from Hb at the tissue site.
- Cyanide combines with methemoglobin to form cyanomethemoglobin, which is non-toxic.
- The study of Hb structure provides an insight into the molecular basis of hemoglobinopathies.
Properties of Hemoglobin
Hemoglobin is a complex protein with several important properties, including:
- Oxygen binding: Hemoglobin can bind with oxygen in the lungs and transport it to tissues throughout the body.
- Allosteric regulation: Hemoglobin undergoes a conformational change upon oxygen binding, allowing for more efficient binding of additional oxygen molecules.
- CO2 transport: Hemoglobin can also bind with carbon dioxide, produced during cellular respiration, and transport it back to the lungs to exhale.
- pH regulation: Hemoglobin can act as a buffer to help maintain the pH balance of the blood.
- Ligand binding: Hemoglobin can also bind with other ligands, such as carbon monoxide, which can lead to poisoning if the levels are too high.
- Structural stability: Hemoglobin is a highly stable protein that can withstand changes in temperature, pressure, and other environmental factors.
- Genetic variability: Hemoglobin has genetic variability across different populations, which can lead to the development of genetic diseases like sickle cell anaemia.
Hemoglobin derivatives
- Hemin (Hematin hydrochloride)
- Hemochromogen
- Hematoid
- Methemoglobin (an oxidation product of Hb; produced by drugs like nitrates, phenacetin, sulphonamide drugs; lack of enzymes like methemoglobin reductase, diaphorase-I, HbM.
- The toxic effects of Met Hb are cyanosis, fatigue, tachycardia, tachypnea, and depression.
- Methemalbumin: (a combination of hematin and albumin), not present in normal adult blood. When present, it indicates intravascular hemolysis, detected by Schumm’s test.
Combination of Hb with gases
- Oxyhemoglobin: (loose and reversible combination with O2); 1.34ml O2 combined with each gm of Hb.
- One mole of Hb can maximally connect with four moles of O2. The partial pressure of O2 favors oxygenation.
- The partial pressure of CO2 favors dissociation. Acidosis favors liberation of O2.
- Oxygenated Hb is relaxed, i.e., in an “R” state. R state is characterized by removing valine residue from the heme pocket of β-subunit with broken salt bridges; it cannot bind BPG; FFe++ comes in the plane of the porphyrin ring; heme-heme interaction increases the affinity for O2; histidines of β-chains release protons (H+).
- Deoxygenated Hb: In “T” form, i.e.., taut form, salt bridges plenty and intact, valine residue covers the heme pocket of β-chain and does not allow entry of O2; can bind BPG; F++ out of the plane of the porphyrin ring; low affinity for O2; β-chain histidine residue protonated (H+ added).
- Carboxyhemoglobin (Hb+ carbo monoxide): Firmer combination, not reversible, the association of Hb to CO is 210 times more than Oxygen; it inhibits cytochrome oxidase of the electron transport chain.
- Carbamino Hb (Hb + CO2):
- Sulfhaemoglobin: Greenish pigment; formed when H2S reacts with Oxy-Hb, seen in severe constipation, certain types of bacteria.
Abnormal Haemoglobins
More than 30 abnormal types descry, differentiated by their characteristic electrophoretic mobilities, generally transmitted; are due to the single mutant gene; Two types –
- Due to the mutation of the structural gene. E.g: HbS, HbM, HbC, HbD (Punjab) etc.
- Due to a mutation in the regulator gene. E.g., Thalassemia.
- Detection by Finger Printing techniques and Hybridization
Effects of abnormal Hb
- Changed Red cell morphology
- Hemolytic anemia, Jaundice
- Methemoglobinemia
- high O2 affinity, e.g., Hb cheaper, Hb-Rainier
- interfering with mRNA formation, e.g., Hb constant spring
HbS
In both β-chains glutamic acid in the 6th position is replaced by valine, resulting in an increase in viscosity and precipitation of HbS. Hence, the crescent or sickle-shaped RBC is of a more fragile nature. However, such RBCs show increased resistance to malaria but are more vulnerable to salmonella infections.
Thalassemias
- α-chain Thalassemia: Synthesis of α-chains is replaced. E.g.: HbH (β4) and Hb-Barts (¥4).
- β-chain Thalassemia (Thalassemia major): The synthesis of β-chain is repressed. As a result, increased synthesis of HbA2 or HbF.
Frequently Asked Questions (FAQs)
What is heme in hemoglobin?
Heme is a crucial component of hemoglobin, the protein that transports oxygen in the blood. Each hemoglobin molecule contains four heme groups, and each heme group contains an iron atom. When oxygen binds to the iron atom, it changes the shape of the hemoglobin molecule, which alters the way the hemoglobin molecule interacts with other molecules in the body. This change in shape allows the hemoglobin molecule to deliver oxygen to the body’s tissues.
Does hemoglobin have beta sheets?
Hemoglobin has two beta strands, and their structure is a little different from that of an alpha helix. The best way to visualize hemoglobin is to make up two alpha-helixes with a loop in between them. There is a histidine amino acid at the end of each strand involved in binding the porphyrin ring.
How is hemoglobin made?
Hemoglobin comprises four protein subunits, which are each composed of a heme group and a globin protein. The heme group contains an iron atom responsible for binding oxygen molecules. The four subunits are held together by noncovalent interactions, which allow the hemoglobin chemical structure to change shape in response to changes in oxygen concentration.
Hemoglobin globular or fibrous?
Hemoglobin is the protein in red blood cells that carries oxygen from the lungs to the rest of the body. It is a globular protein, meaning that it has a round, spherical shape. The hemoglobin molecule comprises four protein subunits, each of which contains heme, a pigment that gives red blood cells their color. When oxygen binds to hemoglobin, it changes the protein’s shape, making it more elongated, and this change in shape helps the hemoglobin molecule transport oxygen more efficiently.
Is hemoglobin composed of DNA?
protein, meaning that it has a round, spherical shape. The hemoglobin molecule comprises four protein subunits, each of which contains heme, a pigment that gives red blood cells their color. When oxygen binds to hemoglobin, it changes the protein’s shape, making it more elongated, and this change in shape helps the hemoglobin molecule transport oxygen more efficiently.
How is hemoglobin synthesized?
Hemoglobin is synthesized in the bone marrow in a process that involves multiple steps. First, the marrow produces erythroblasts, which are immature red blood cells. These cells then mature and begin to produce hemoglobin. The hemoglobin molecule consists of a protein called heme and a small molecule called globin. The heme portion of the molecule gives hemoglobin its red color, while the globin portion consists of two proteins, alpha, and beta-globin. The alpha and beta globins combine to form a structure that resembles a spiral staircase. This structure is what allows hemoglobin to bind to oxygen molecules.
What is the hemoglobin formula?
Hemoglobin is a protein in red blood cells that carries oxygen from the lungs to the rest of the body. The hemoglobin formula is C29H46N4O9. The structure of hemoglobin is a tetramer, meaning it comprises four subunits, and each subunit contains an iron atom responsible for binding oxygen.
What is the chemical makeup of Hemoglobin?
The chemical makeup of hemoglobin can be illustrated by its molecular structure.
- Hemoglobin is composed of four protein subunits, each of which contains a heme group with an iron atom in the center.
- The iron atom is responsible for binding to oxygen molecules and allowing for the transport of oxygen in the bloodstream.
- The molecular structure of hemoglobin can be visualized as a globular protein with a quaternary structure, where each subunit is represented by a different color.
- The heme group, which contains the iron atom, is located within each subunit and is represented by the red, blue, green, and yellow spheres.
- The molecular structure of hemoglobin is critical to its function in transporting oxygen and other molecules throughout the body.
Final words
Hemoglobin is a complex protein molecule that is found in red blood cells. It is composed of heme groups, which are made up of porphyrin rings and amino acids.
Hemoglobin is responsible for transporting oxygen and carbon dioxide through the bloodstream.
Hemoglobin comprises four subunits: two alpha and two beta. This blog will explain the structure of hemoglobin and how it works.
Red blood cells are essential in our lives, and they provide oxygen to our tissues and organs.
Hemoglobin is the main component of red blood cells, and they help carry oxygen throughout the body and regulate the amount of oxygen supplied to tissues.