In the United States, each year about 1 in 10,000 to 15,000 babies is born with PKU. This disease occurs in all ethnic groups. But it is most common in Native Americans and northern Europeans in African Americans, or Japanese in Ashkenazi Jews.

What is Phenylketonuria?
Phenylketonuria (PKU for short) is a condition in which the body cannot process an amino acid called phenylalanine. Amino acids help build protein in the body. Without treatment, phenylalanine builds up in the blood and causes health problems. Phenylketonuria is one of the most common recessive genetic disorders in humans. PKU is an inborn error of metabolism.
What causes PKU?
PKU is inherited. That means they pass it from parents to children through genes. A gene is a part of the cells in your body that contains instructions on how your body grows and functions. Genes come in pairs; each person inherits a gene pair from their father and a gene pair from their mother. The genotypic frequency of an inherited autosomal recessive condition, phenylketonuria, is 1 in 36000. Chegg investigated phenylketonuria, why individuals with this condition are sensitive to aspartame.
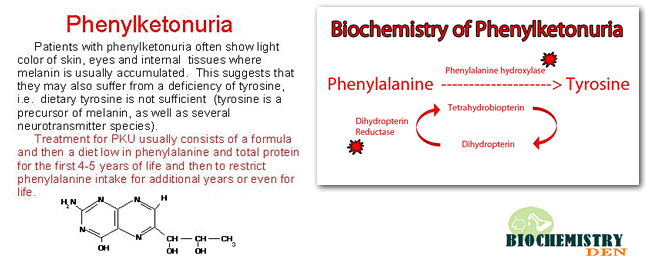
Sometimes genes can undergo a sudden change. It calls this change “gene mutation.” Parents can pass on the changes in genes to their children. Sometimes changes can cause the gene to not function properly. Sometimes it can cause birth defects or other health problems.
A birth defect is a health problem that is present when the baby is born. To have PKU, your child must inherit a change in the gene for PKU from both parents. If you inherit the gene change, only one parent has the gene change for PKU but does not have the disease. In that case, she tells her baby she is a carrier of PKU. The carrier has a PKU gene change but does not have the disease.
How to tell if your baby has PKU?
All babies are doing screening tests for newborns for phenylketonuria. Screening for newborns detects conditions in the baby at birth; they are rare but serious. These include a blood test, hearing, and heart. With screening, PKU can be detected and treated early, so babies can grow healthy. Before giving your baby high, the health professional will take a few drops of blood from the heel. They collected the blood on a special paper and sent it to a laboratory for analysis. The lab sends the test results to health care for your baby.
- Estimation of Blood Glucose level by Folin-Wu method
- What is Paper Chromatography? Principle and Procedure
If the results are not normal, it means he must do more tests on your baby. The health professional may recommend another type of test, called a diagnostic test, instead. This test can check if your baby has phenylketonuria or if abnormal results are because of other causes. If you are doing the test on your baby before their first full day of life, it may not detect PKU. Some experts recommend that if you do the test on your baby within 24 hours of life, the test has to be repeated at 1-2 weeks of age.
Phenylketonuria symptoms
What problems can cause PKU?
Babies born with phenylketonuria appear normal for the first few months of life. But without treatment, they have signs and symptoms of the disease at about 6 months of age. Phenylketonuria mutation affects protein changes in amino acids, DNA, RNA, and amino acids.
These include:
- Jerky movements of the arms and legs
- Skin and eyes are lighter colors. Infants with PKU cannot adequately produce melanin, the pigment responsible for skin color and hair.
- Body odor similar to rust
- Seizures
- Skin Rashes
- Head size: small
- Learn to sit, crawl, or walk later than planned.
- loses interest in the surrounding environment
- Delays in mental and social skills
- Intellectual disabilities
- Behavioral problems such as hyperactivity
Phenylketonuria treatment
If, your baby, has PKU, what kind of treatment you need?
You may need weekly or more frequent testing during the first year of life to control their phenylalanine levels. After that, you may get tested once or twice a month during childhood.
Your baby should follow a particular meal plan low in phenylalanine. It is best to start this plan as soon as possible, ideally within the first 7–10 days of life. First, it gives your baby a unique formula of protein with reduced phenylalanine. Protein is essential to help your baby grow and develop. It controls the amount of phenylalanine in the formula to meet the individual needs of your baby. It can also give him some milk. Breast milk contains phenylalanine, so talk to your health professional to find out how much your baby’s breast milk can provide.
When your baby is ready to eat solid foods, you can eat vegetables, fruits, some grains (such as cereals, bread, and pasta, which are low in protein), and other foods low in phenylalanine. If your baby has PKU, you should not eat:
Milk, cheese, ice cream, or other dairy products
- Eggs
- Meat or poultry
- Fish
- Nuts
- Beans
- Food or beverages with aspartame, an artificial sweetener that contains much phenylalanine.
Meal plans for PKU are different for each baby and will vary over time depending on the amount of phenylalanine that the baby can process. The health professionals at a medical center or clinic with a special program to treat PKU can help you create a meal plan for your baby. Ask for information on the professional health of your baby at the medical centers or clinics that treat PKU.
Your child should follow the meal plan for PKU’s entire life. If your child becomes pregnant, follow the meal plan during the pregnancy. Most pregnant women with PKU can have healthy pregnancies and babies.
The drug Kuvan (sapropterin dihydrochloride) may help people with PKU. It is more likely to take effect in people with mild PKU special forms. Children taking Kuvan must follow a special meal plan, but possibly not as strict as those not taking the drug. Anyway, still, they need frequent blood tests to check levels of phenylalanine.
Source: PKU